توسط بهپور ریاحی پور
 ذخیره PDF
ذخیره PDF چاپ نوشته
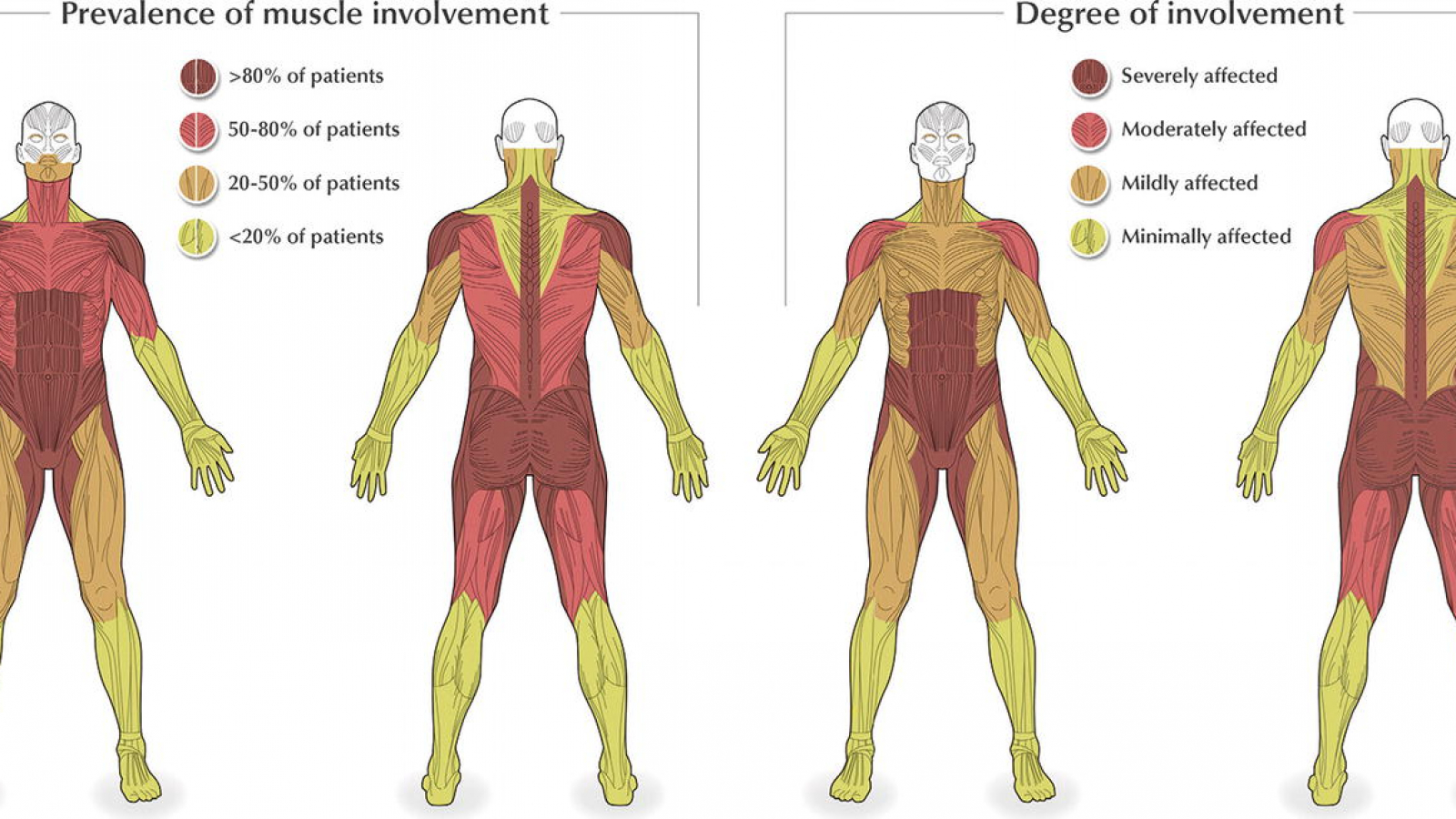
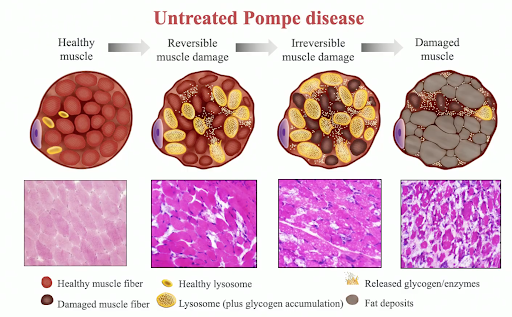
چاپ نوشتهبیماری پمپه یک اختلال ژنتیکی نادراست که در آن بدن توانایی شکستن قندهای پیچیده (گلیکوژن) را ندارد که بروی بافت ها و ارگان ها مخصوصا عضلات اثر دارد.
این بیماری نتیجه کمبود نقص یک آنزیم به نام گلوکوزیداز اسید آلفا که قندهای پیچیده را در بدن می شکند، می باشد. موتاسیون در ژن GAA که در روند شکستن گلیکوژن نقش دارد باعث این اختلال می گردد.
این بیماری به سه دسته تقسیم می شود:
نشانه های کلاسیک این بیماری در نوزادان چندماه بعد از تولد بروز پیدا می کند.
نشانه های غیرکلاسیک در حدود یک سالگی بروز می کند.
نشانه های دیررس در زندگی کودک یا حتی در سنین نوجوانی یا بزرگسالی ظاهر می شود.
چه کسانی درگیر بیماری پمپه می شوند:
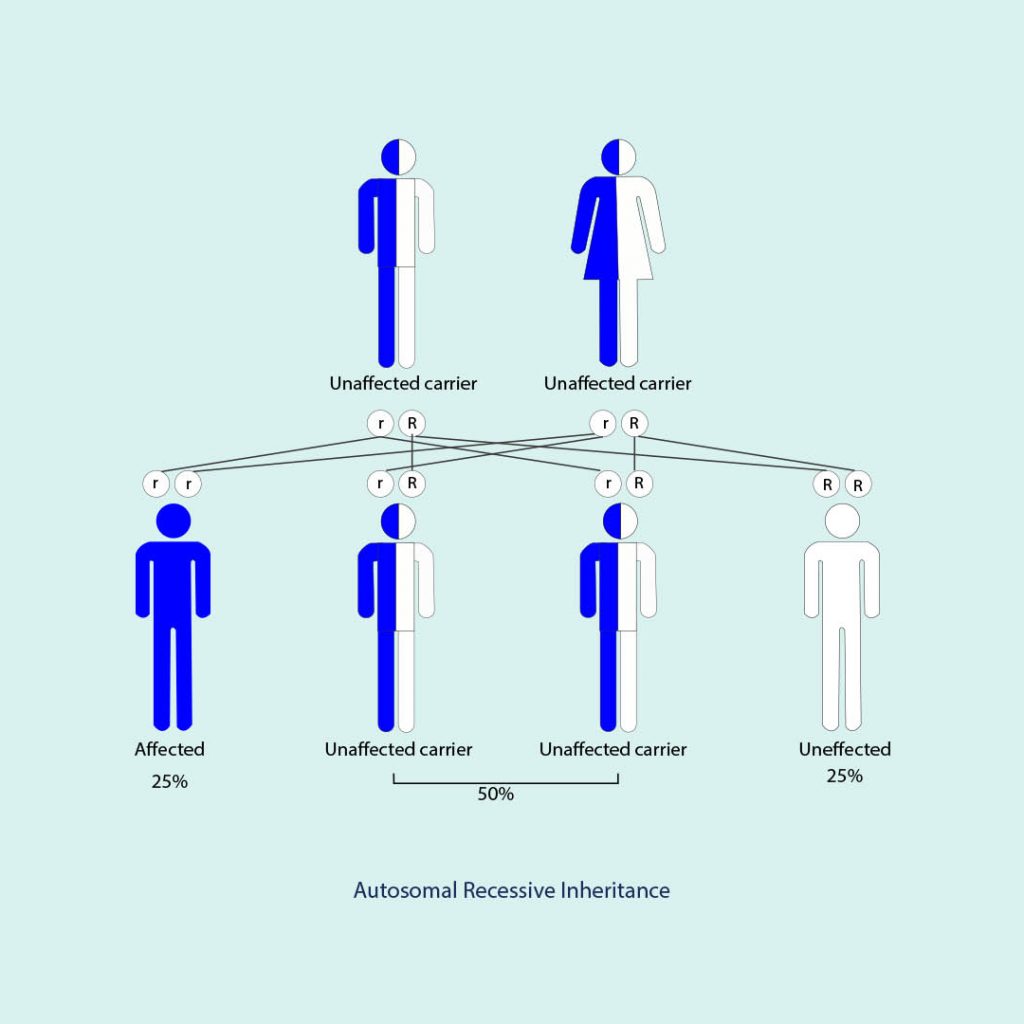
از آنجایی که این یک بیماری ژنتیکی می باشد، فردی که درگیر این بیماری می شود از والدین خود به ارث می برد. این بسیار متداول است که والدین هیچ علامت ونشانه ای را بروز ندهند.
این بیماری بسیار نادر است . در ایالت متحده امریکا یک نفر از هر 40000 نفر با این بیماری درگیر می شود و شیوع آن بین همه ی گروه های نژادی یکسان است.
علائم و علت ها
نشانه ها می تواند متفاوت باشد بسته به اینکه بیماری در چه سنی خود را بروز دهد. در نوزادان علائم کلاسیک شامل ضعف عضلات، افزایش اندازه کبد، کاهش وزن و عدم افزایش وزن و رشد با سرعت مورد انتظار، تنفس مشکل دار، مشکلات تغذیه ای، عفونت در سیستم تنفسی، مشکلات شنوایی و علایم غیرکلاسیک شامل تاخیر در مهارت های غیرحرکتی مانند چرخیدن و نشستن، ضعف تدریجی عضلات، افزایش غیرطبیعی اندازه قلب ، تنفس مشکل دار می باشد.
علائمی که در افراد بالغ گزارش شده است:
ضعف تدریجی در پاها و تنه ، مشکلات تنفسی، قلب بزرگ شده ، به سختی راه رفتن، درد عضله در مناطق وسیعی از بدن. از دست دادن توانایی ورزش کردن، اغلب زمین خوردن، عفونت های متداول ریوی، سردردهای صبحگاهی، خستگی در طول روز. از دست دادن وزن بدن، مشکلات بلع، ضربان نامنظم قلبی، مشکلات شنوایی. سطوح افزایش یافته کراتین کیناز(CK) آنزیمی که به عملکرد بدن بسیار کمک می کند و باعث بهبود سطح انرژی به سلول ها می شود.
تشخیص و آزمایشات
نمونه خون از فرد گرفته می شود و آنزیم ذکر شده مورد ارزیابی قرار می گیرد. همچنین تست های تنفسی برای اندازه گیری ظرفیت ریه ها و الکترومیوگرافی. (آزمایشی که اندازه گیری می کند آیا عضلات به خوبی کار می کنند یا نه؟). سپس تایید نتایج با استفاده از تست DNA انجام می گردد.
همچنین گرفتن سابقه و تاریخچه خانوادگی کامل بیمار، تست های تنفسی برای اندازه گیری ظرفیت ریه ها، الکترومیوگرافی و MRI، بررسی ضربان قلب با استفاده از X-ray ، الکتروکاردیوگرام و اکوکاردیوگرام، مطالعه و بررسی وضعیت خواب فرد بسیار با اهمیت است.
تشخیص پیش از تولد برای مادران با ریسک انتقال این بیماری انجام می شود.
مدیریت بیماری و درمان
درمان جایگزیمی آنزیمی(ERT ) یک درمان ثابت شده برای همه بیماران پمپه می باشد. داروی گلوکوزیداز آلفا دارویی است که به صورت داخل رگی تزریق می گردد. این آنزیم با روش مهندسی ژنتیک تولید شده است. که به صورت طبیعی مانند آنزیم گلوکوزیدار اسید آلفا عمل می کند.
تیم متخصص (پزشکان قلب و دستگاه تنفسی، نورولوژیست ها و غیره) می توانند علائم و نشانه های فرد را کنترل کنند و مراقبت های حمایت کننده ای برای بیماران پمپه درنظر بگیرند.
بدون درمان، نوزادان با بیماری پمپه فوت خواهند کرد. بسیاری از افراد با بیماری پمپه دارای مشکلات تنفسی، قلبی و تقریبا همه با ضعف عضلانی درگیر می شوند. بیشتر افراد مجبور به استفاده از کپسول اکسیژن و ویلچیر می شوند.
پیشگیری
به دلیل اینکه این بیماری ژنتیکی می باشد نمی توان آن را به طور کامل پیشگیری نمود. فقط می توان با درمان های حمایتی، علائم آن را کنترل کرد.
بیماران با هر کدام از انواع بیماری پمپه با تشخیص به موقع می توانند زندگی طولانی تری داشته باشند. بنابراین، همه انواع این بیماری اغلب کشنده می باشد. بیماران درگیر با نوع شروع زودرس دوران نوزادی غیرکلاسیک ممکن است تا دوران اولیه کودکی زنده بمانن. بچه هایی با نوع شروع دیر رس بیماری می توانند طولانی تر زنده بمانند و پیشرفت بیماری بسیار آهسته پیش رود.
تهیه و ترجمه توسط : خانم شراره سلمانی زاده ( آزمایشگاه ژنتیک پزشکی ژنوم اصفهان – مرکز تحقیقات سلولی، مولکولی و ژنتیک ژنوم اصفهان ).




 آزمایشگاه ژنتیک در اصفهان | ژنتیک در اصفهان |مشاوره ژنتیک در اصفهان | غربالگری | بهترین آزمایشگاه ژنتیک در اصفهان | آزمایشاه ژنتیک سرطان اصفهان
آزمایشگاه ژنتیک در اصفهان | ژنتیک در اصفهان |مشاوره ژنتیک در اصفهان | غربالگری | بهترین آزمایشگاه ژنتیک در اصفهان | آزمایشاه ژنتیک سرطان اصفهان